At present, my research is focused on computer simulation of soft and biological matter. The focus is on developing and applying innovative simulation, modeling, and theoretical approaches to study complex molecular systems.
Soft and biological matter is characterized by physical properties that are determined by the interplay of disparate length and time scales. Multiscale simulation techniques that couple multiple models at different resolutions provide the most efficient way to span many orders of magnitude in the spatiotemporal scales involved in these systems.
AdResS: Adaptive Resolution Simulation
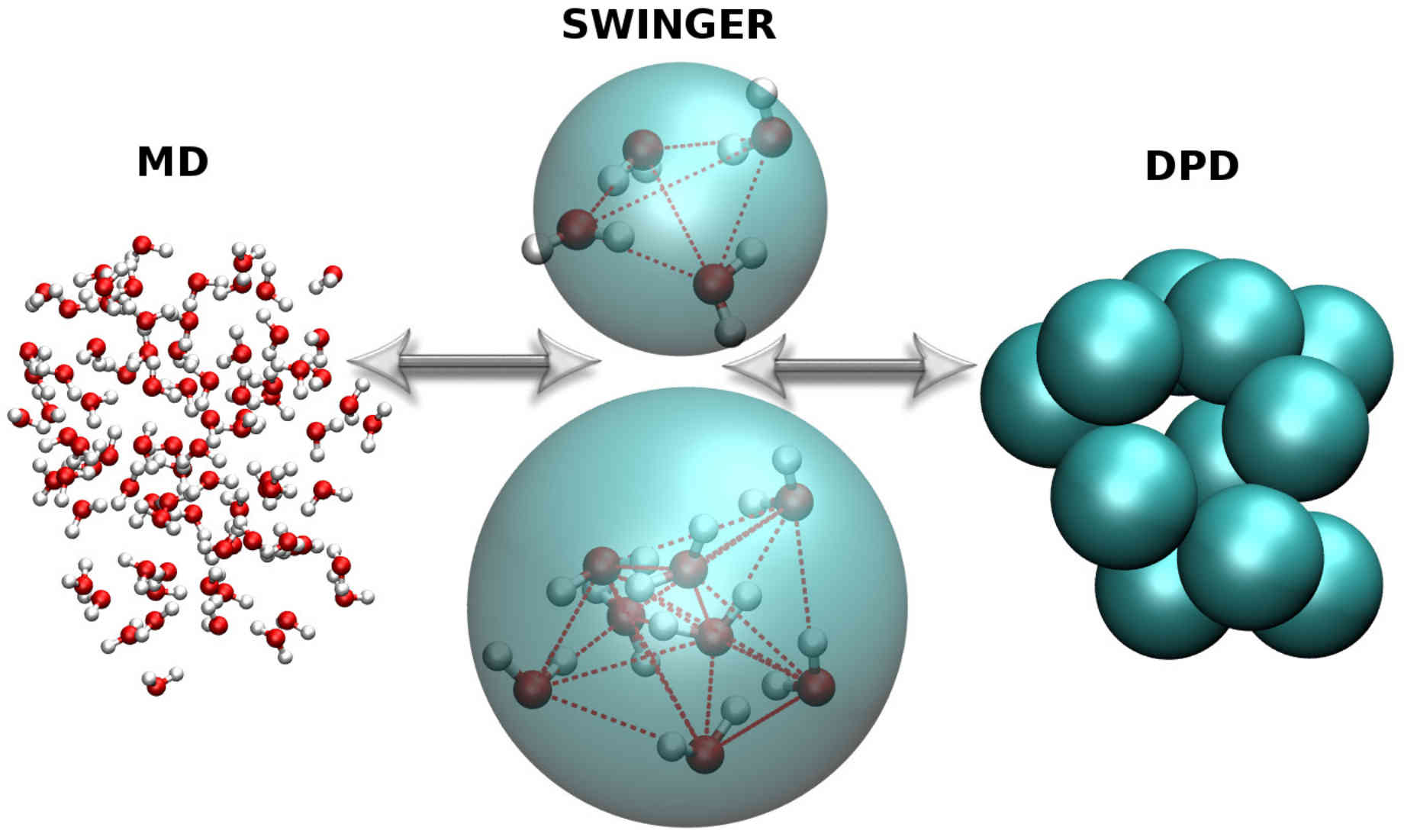
SWINGER: Dynamic Clustering Algorithm
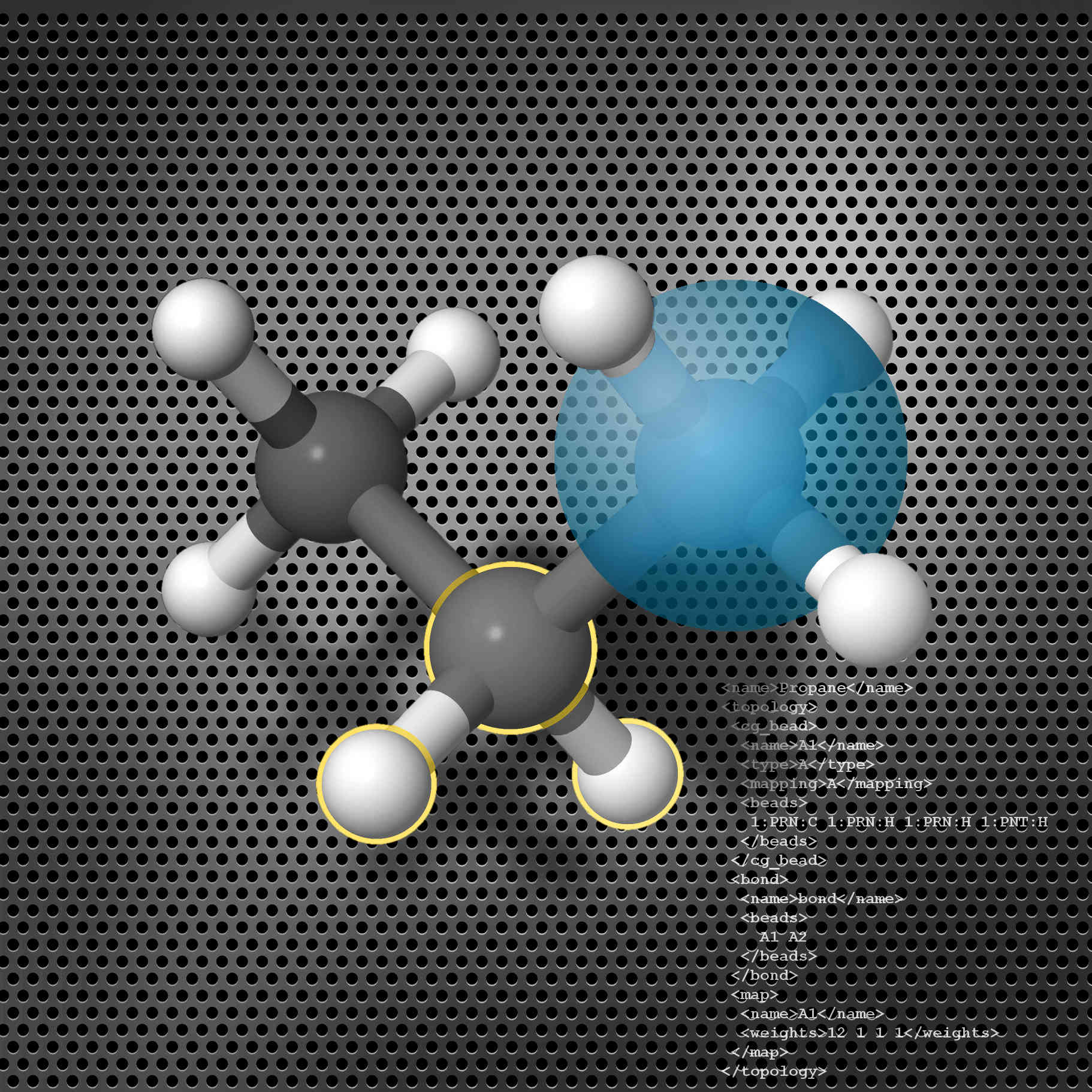
STOCK: Structure Mapper and Online Coarse-Graining Kit for Molecular Simulations
Coupling Atomistic and Continuum Hydrodynamics
Open Boundary Molecular Dynamics
Open molecular systems exchange mass, momentum, and energy with their surroundings. Our OBMD methodology enables equilibrium molecular dynamics simulations in the grand-canonical ensemble as well as nonequilibrium fluid flow simulations. The flow is introduced as an external boundary condition while the equations of motion for the bulk remain unaltered.
Nanofluidics
We simulate fluid flows past and through nano-objects using computational fluid dynamics aiming to reproduce essential features of the fluid flows computed by molecular dynamics. Computational advantages of computational fluid dynamics allow for simulations well beyond the current reach of molecular dynamics. Computational results are supplemented by analytical calculations.
ERC AdG MULTraSonicA
MULTraSonicA aims to design a virtual research environment to assist medical applications of ultrasound-guided drug and gene delivery and imaging. The project focus are the encapsulated microbubbles and gas vesicles with submicron size that are used as ultrasound contrast agents and can also act as drug carriers. We will develop new, data-informed mesoscopic models of ultrasound contrast agents to accurately model their rheological and acoustic behavior that critically affects the technology of ultrasound-guided drug and gene delivery. Interactions of ultrasound and agents at a submicron level will be included by harnessing high-performance computing (HPC) and employing novel multiscale methods that enable seamless propagation of ultrasound from the macro to microscopic level. The proposed framework will be integrated with experimental efforts to advance ultrasound-guided drug and gene delivery across biomedical applications.
MultiXscale – EuroHPC JU Center of Excellence
MultiXscale involves 16 consortium partners who jointly develop multiscale simulation software to solve societal challenges associated with biomedicine, the transition to sustainable energy, and civil transport by supercomputers. The goal of MultiXscale is to develop software for computationally efficient multiscale simulations running on (pre)exascale HPC systems. The computer simulation codes and developed multiscale workflows will be openly accessible to researchers and engineers, user-friendly, easy-to-install, and with sustainable user support.
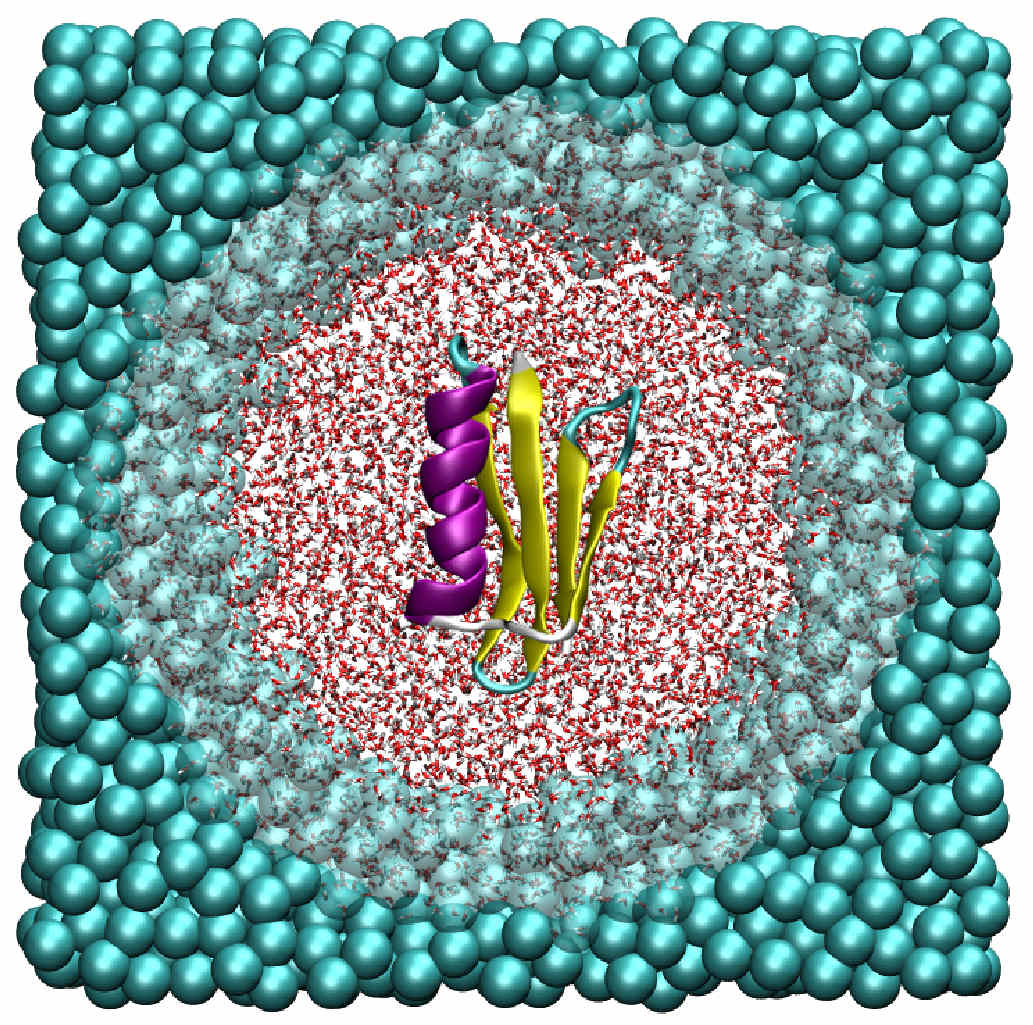
Adaptive resolution simulation of an atomistic protein in MARTINI water
We couple atomistic water around the protein with mesoscopic water, where four water molecules are represented with one coarse-grained bead, farther away. The water molecules change their resolution from four molecules to one coarse-grained particle and vice versa adaptively on-the-fly.
Our multiscale model is compatible with the widely used MARTINI force field and will therefore significantly enhance the scope of biomolecular simulations.
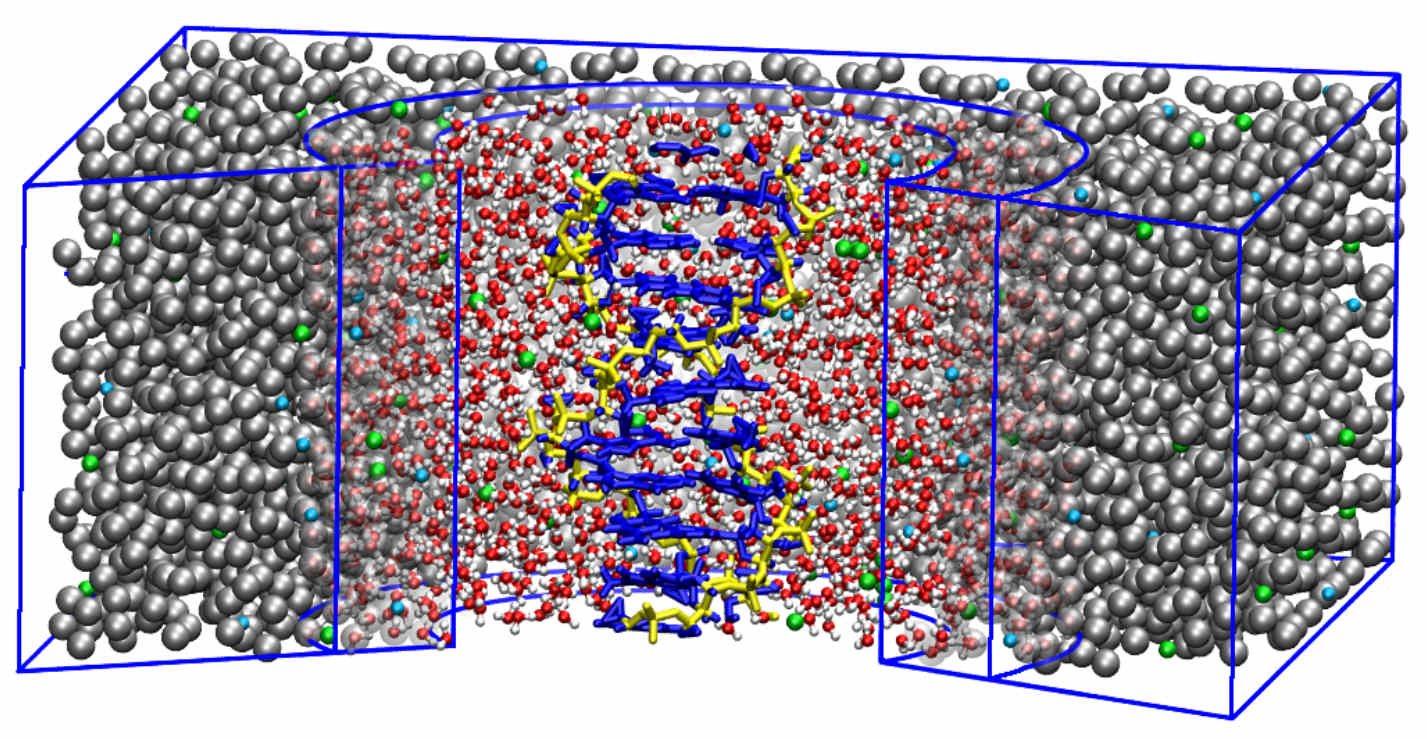
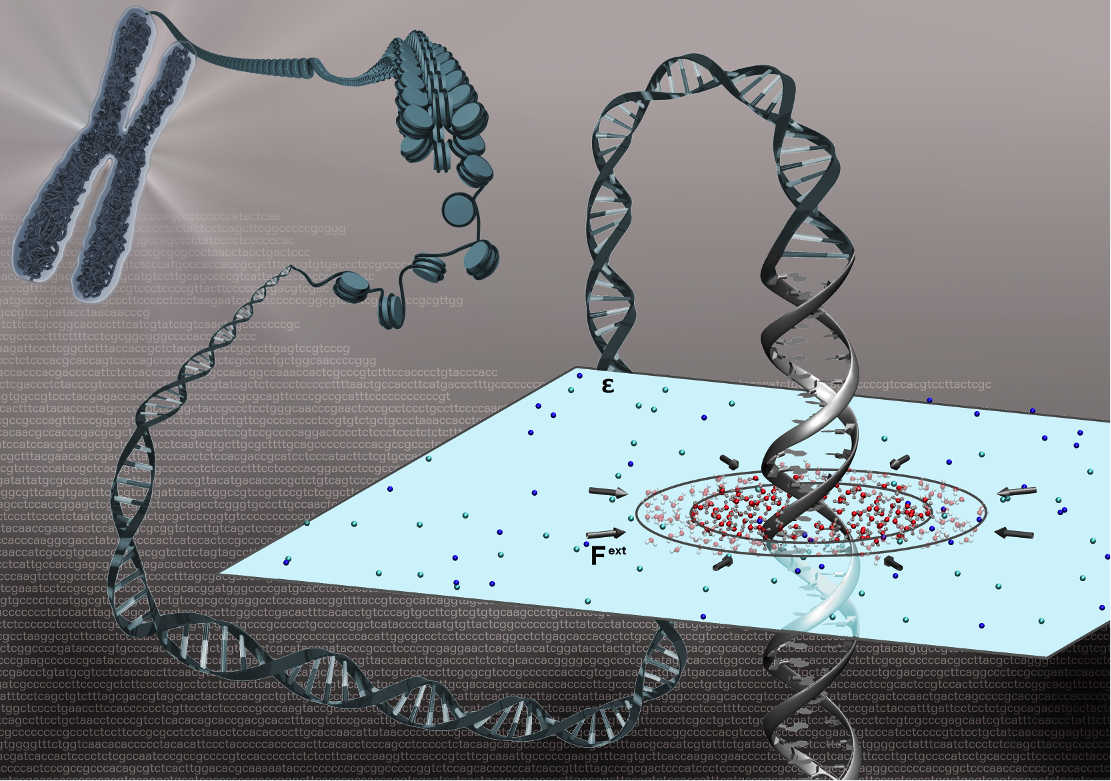
Adaptive resolution simulation of a DNA molecule in salt solution
We present a multiscale simulation of a DNA molecule in 1 M NaCl salt solution environment, employing the adaptive resolution simulation approach that allows the solvent molecules, i.e., water and ions, to change their resolution from atomistic to coarse-grained and vice versa adaptively on-the-fly.
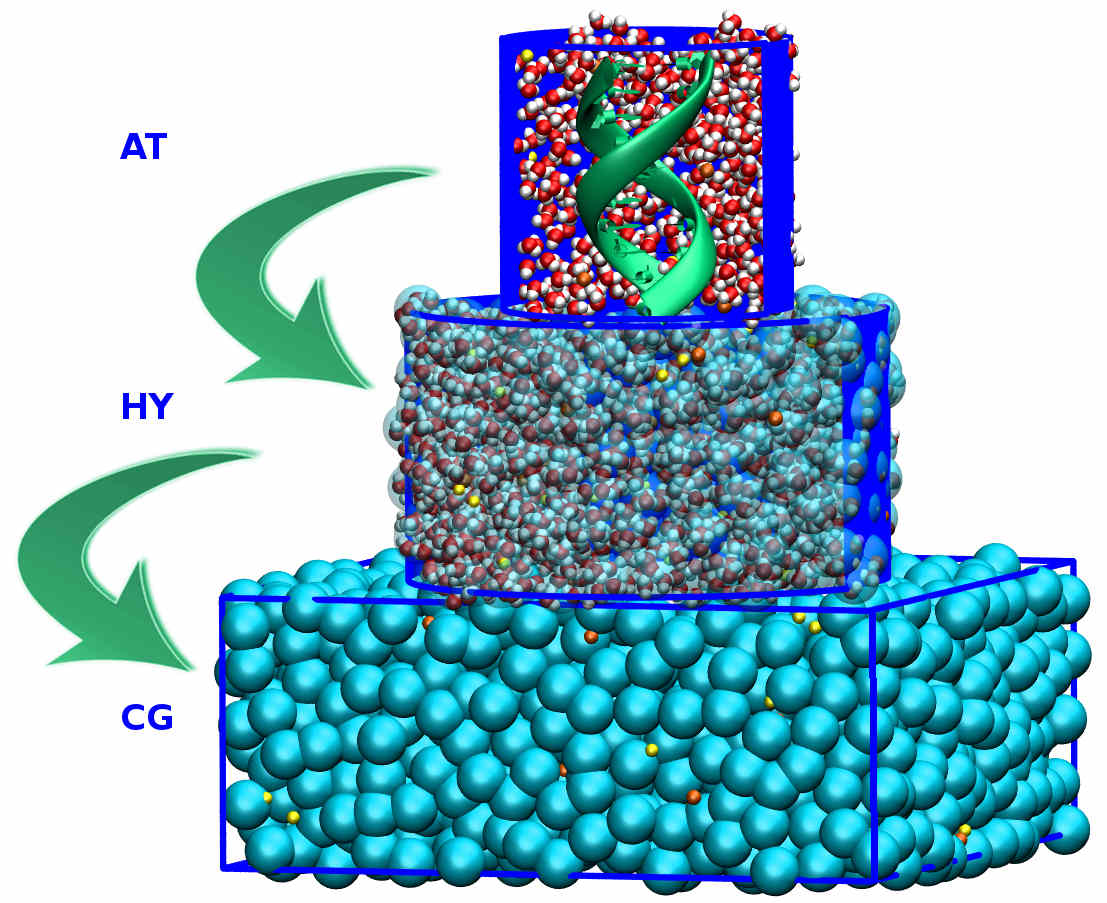
Adaptive resolution simulation of a DNA molecule in MARTINI salt solution
We present a dual-resolution model of a DNA molecule in a bathing solution, where we concurrently couple atomistic bundled water and ions with the coarse-grained MARTINI model of the solvent.
Article:
Eur. Phys. J. Special Topics, DOI: 10.1140/epjst/e2016-60117-8
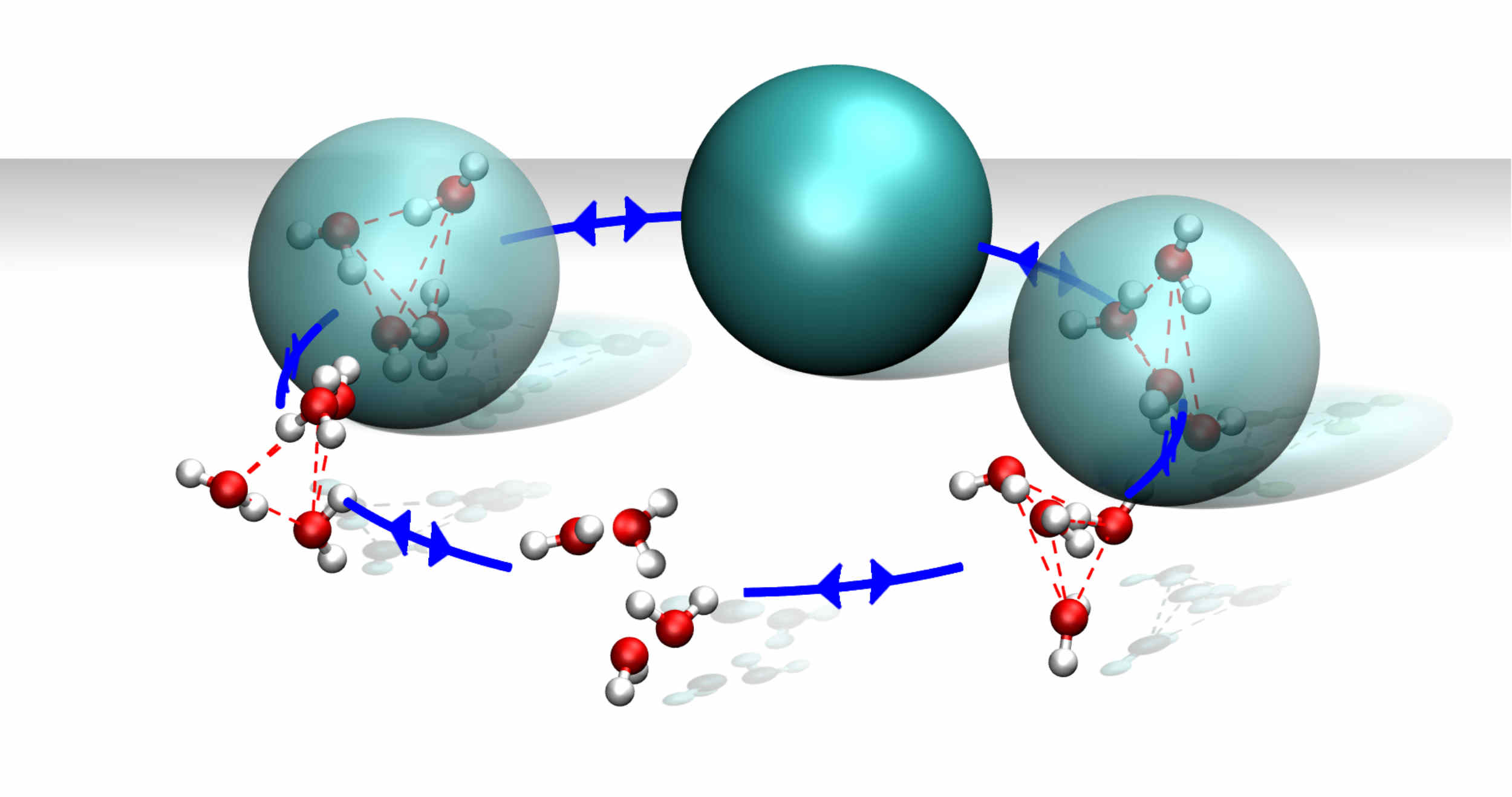
SWINGER is a dynamic clustering algorithm that can concurrently assemble, disassemble, and reassemble water bundles, consisting of several water molecules. Thus, it allows for a seamless coupling between standard atomistic and supramolecular water models in adaptive resolution simulations.
SWINGER paves the way for efficient multiscale simulations of biomolecular systems without compromising the accuracy of atomistic water models.
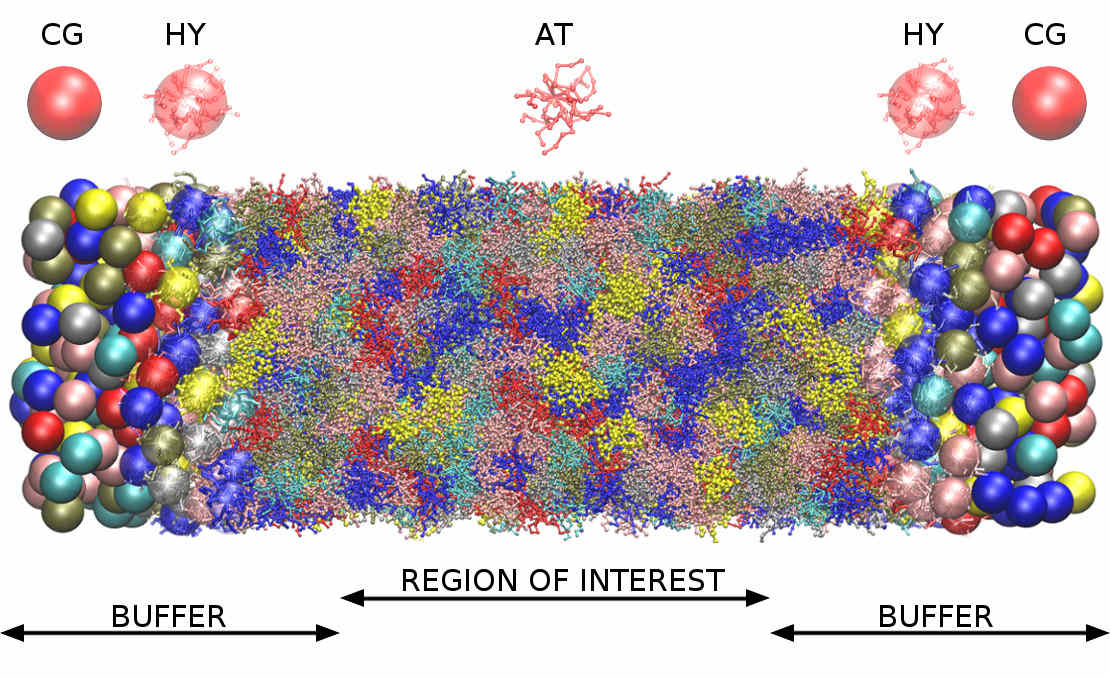
The core domain of the simulation box is solved using the all-atom resolution. The coarse-grained layer introduced by AdResS is located at the outer part of the open simulation box to enable the insertion of large molecules into the system.
In the explicit domain, the water molecules and ions are both overtly present in the system, whereas in the implicit water domain, only the ions are explicitly present and the water is described as a continuous dielectric medium. Water molecules are inserted and deleted into/from the system in the intermediate buffer domain that acts as a water reservoir to the explicit domain, with both water molecules and ions free to enter or leave the explicit domain.
Our approach is general and allows for efficient molecular simulations of biomolecules solvated in bathing salt solutions at any ionic strength condition.
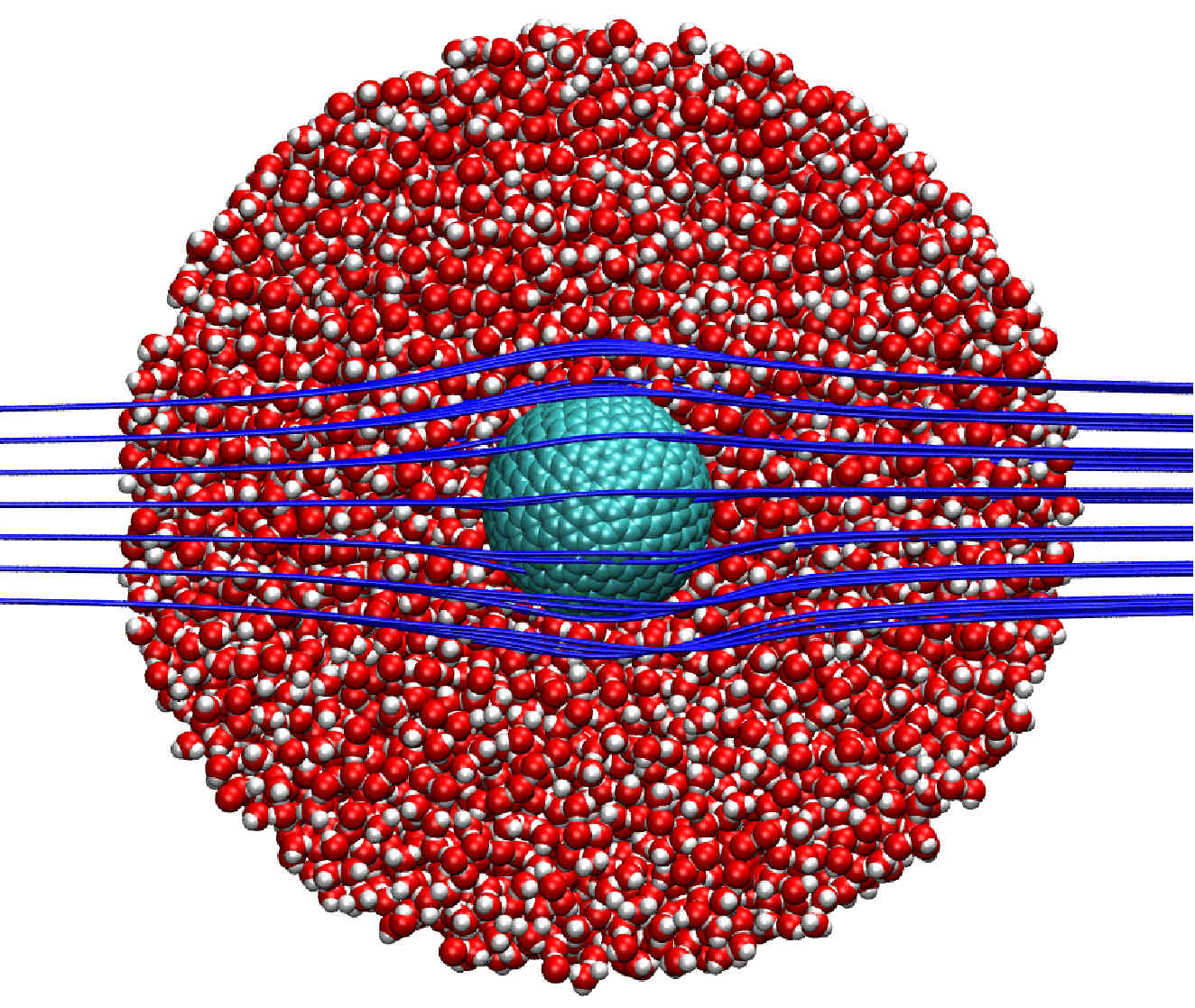
Continuum simulations of water flow in carbon nanotube membranes
We propose the use of the Navier–Stokes equations subject to partial-slip boundary conditions to simulate water flows in Carbon NanoTube (CNT) membranes.